- Publications
- Protocols
- Software
- Soybean IVT Array
- Soybean Whole Genome Array
- Arabidopsis ATH1 Array
- Presentations & Videos
- People
Publications
Jo, L., Pelletier, J.M., Hsu, S., Baden, R., Goldberg, R.B., Harada, J.J. (2020) Combinatorial interactions of the LEC1 transcription factor specify diverse developmental programs during soybean seed development, Proc. Natl. Acad. Sci. USA, 117:2, 1223-1232. [DOWNLOAD PDF]
Chen, M., Bui, A.Q., & Goldberg, R.B. (2020) Using giant scarlet runner bean (Phaseolus coccineus) embryos to dissect the early events in plant embryogenesis. in Plant embryogenesis: Methods and Protocols , M. Bayer, Ed. (Springer Science+Business Media, 2020), 2122, 205-222. [DOWNLOAD PDF]
Jo, L., Pelletier, J.M., Harada, J.J. (2019) Central role of the LEAFY COTYLEDON1
transcription factor in seed development, Journal of Integrative Plant Biology 61:5, 564-580. [DOWNLOAD PDF]
Chen, M., Lin, J., Hur, J., Pelletier, J.M., Baden, R., Pellegrini, M., Harada, J.J. and Goldberg, R.B. (2018). Seed genome hypomethylated regions are enriched in transcription factor genes. Proc. Natl. Acad. Sci. USA. 115, E8315-E8322. [DOWNLOAD PDF]
Henry, K.F., Bui, A.Q., Kawashima, T., and Goldberg, R.B. (2018). A shared cis-regulatory module activates transcription in the suspensor of plant embryos. Proc. Natl. Acad, Sci. USA. (2018). 115 (25) E5824-5833. [DOWNLOAD PDF]
Pelletier, J.M., Kwong, R. Park, S., Le, B.H., Baden, R., Cagliari, A., Hashimoto, M., Munoz, M., Fischer, R.L., Goldberg, R.B., Harada J.J. (2017) LEC1 sequentially regulates the transcription of genes involved in diverse developmental processes during seed development. Proc. Natl. Acad. Sci. USA, 114:32, E6710–E6719. [DOWNLOAD PDF]
Lin. J., Le, B.H., Min, C., Henry, K.F., Hur, J., Hsieh, T-F., Chen, P-Y., Pelletier, J.M., Pellegrini, M., Fischer, R.L., Harada, J.J., and Goldberg, R.B. (2017). Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA. 114, E9730-E9739. [DOWNLOAD PDF]
Danzer, J., Mellott, E., Bui, A.Q., Le, B.H., Martin, P., Hashimoto, M., Perez-Lesher, J., Chen, M., Pelletier, J.M., Somers, D.A., Goldberg, R.B., Harada, J.J. (2015) Down-regulating the expression of 53 soybean transcription factor genes uncovers a role for SPEECHLESS in initiating stomatal cell lineages during embryo development, Plant Physiology, 168:3, 1025-35. [DOWNLOAD PDF]
Henry, K.F., Kawashima, T., and Goldberg, R.B. (2015). A cis-regulatory module activating transcription in the suspensor contains five cis-regulatory elements. Plant Mol. Biol. 88, 207-217. [DOWNLOAD PDF]
Henry, K.F. and Goldberg, R.B. (2015). Using giant scarlet runner bean embryos to uncover regulatory networks controlling suspensor gene activity. Frontiers in Plant Science 6, 44, 1-6. [DOWNLOAD PDF]
Belmonte, M., Kirkbride, R.C., Stone, S.L., Pelletier, J.M., Bui, A.Q., Yeung, E.C., Hashimoto, M., Fei, J., Harada, C.M., Munoz, M.D., Le, B.H., Drews, G.N., Brady, S.M., Goldberg, R.B., and Harada, J.J. (2013) Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc. Natl. Acad. Sci. USA, 110:5, E434-44. [DOWNLOAD PDF]
Le, B.H., Cheng, C., Bui, A.Q., Wagmeister, J.A., Henry, K.F., Pelletier, J., Kwong, L., Belmonte, M., Kirkbride, R., Horvath, S., Drews, G.N., Fischer, R.L., Okamuro, J.K., Harada, J.J., and Goldberg, R.B. (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci USA, 170:18, 8063-70. [DOWNLOAD PDF]
Kawashima, T., Wang, X., Henry, K.F., Bi, Y., Weterings, K., and Goldberg, R.B. (2009). Identification of cis-regulatory sequences that activate transcription in the suspensor of plant embryos. Proc. Natl. Acad. Sci. USA, 106, 3627-3632. [DOWNLOAD PDF]
Le, B. H., Wagmaister, J.A., Kawashima, T., Bui, A. Q., Harada, J.J., and Goldberg, R.B. (2007). Using genomics to study legume development. Plant Physiology 144, 562-584 (doi.org/10.1104/pp.107.100362). [DOWNLOAD PDF]
Weterings, K., Apuya, N.R., Bi, Y., Fischer, R.L., Harada, J.J., and Goldberg, R.B. (2001). Regional localization of suspensor mRNAs during early embryo development. Plant Cell, 13, 2409-2425. [DOWNLOAD PDF]
Protocols Used in The Project
Please click on the protocol name to download the pdf file.
Tissue Collection, Fixation, Embedding and Sectioning
- Soybean Seed Fixation and Embedding Protocol
- Arabidopsis Tissue Harvest & Fixation Protocol
- Sectioning Protocol For LCM Capturing
- Hand dissect Scarlet Runner Bean Embryos
Laser Capture Microdissection (LCM)
RNA Isolation From LCM Captured Tissues
- Soybean RNA Isolation Protocol with Arcturus PicoPure Kit
- Arabidopsis RNA Isolation Protocol with Ambion RNAqueous Micro Kit
DNA Isolation From LCM Captured Tissues
Soybean Chromatin Immunoprecipitation (ChIP) Protocol
Illumina Sequencing Library Construction
- RNA-Seq Protocol
- Bisulfite Converted Genomic DNA Library Construction Protocol
- Small RNA-Seq Protocol
GeneChip Hybridization
- Soybean RNA Amplification and cRNA Labeling Protocol
- Soybean- Affymetrix GeneChip Hybridization with Enzo Labeled cRNA
- Arabidopsis Pico RNA Amplification Protocol (NUGEN WTA)
- Arabidopsis Fragmentation and Labeling Protocol (NUGEN Encore)
- Arabidopsis Affymetrix GeneChip Hybridization with NUGEN Labeled cDNA
qRT-PCR
Software
This section provides software used or developed for the analysis of large datasets.
ChipEnrich
Click here to download the latest version (Chipenrich-1.42a.jar)
We modified the ChipEnrich software program (Orlando et al., 2009) to identify GO terms, metabolic pathways, transcription factor families, and DNA sequence motifs overrepresented in coexpressed gene sets and to discover potential transcriptional modules,. This Java program was developed originally to identify significantly enriched GO terms (2009 download) and transcription factor families from gene lists. Significance of enrichment is reported as p values calculated from the hypergeometric distribution (Gadbury et al., 2009) using the Apache Commons Math library (http://jakarta.apache.org/commons/math). The following functions were added to ChipEnrich:
Metabolic pathway enrichment analysis: Genes represented on the ATH1 GeneChip were annotated according to metabolic pathways described in the PATHWAYS database from AraCyc (http://www.arabidopsis.org/biocyc/index.jsp; 2008 download). Enrichment was defined as the ratio of (i) the number of AGI locus identifiers in the query list annotated as belonging to a pathway to (ii) the number of AGI locus identifiers associated with the pathway in the GeneChip compared with the ratio of (iii) the total number of AGI locus identifiers present in the query list to (iv) the total number of AGI locus identifiers present on the GeneChip.
DNA motif enrichment analysis: Gene sets were analyzed to identify enriched DNA sequence motifs known to interact with TFs (Arabidopsis Gene Regulation Information Server, http://arabidopsis.med.ohio-state.edu/, August, 2009) that are located in the region 1 kb upstream of the gene's transcription start site (TAIR9, www.arabidopsis.org) as described by others (O'Connor et al., 2005; Vandepoele et al., 2009). The background distribution was determined by identifying DNA motifs for all genes represented as singletons on the Arabidopsis ATH1 GeneChip. Statistical enrichment (p value < 0.001) was determined for each gene list using the hypergeometric distribution. Enriched DNA sequence motifs are also identified among genes overrepresented for a GO term within a gene list.
Putative Transcriptional Modules: To discover putative transcriptional modules, we associated significantly enriched DNA sequence motifs with transcription factors known or predicted to bind the motifs. We used known interactions between transcription factors and DNA motifs specified in AtcisDB (Davuluri et al., 2003) and defined by others in the literature and assumed that transcription factors of a particular family bind to the same DNA motif (Brady et al., 2007). Two variations of this approach were used. In the first approach, we associated DNA motifs significantly enriched within a coexpressed gene set with their cognate TFs that were included in the coexpressed gene set. In the second, we identified DNA motifs that were significantly enriched for genes corresponding to an overrepresented GO term and associated coexpressed TFs known or predicted to bind the enriched DNA motifs. Using the software package, ChipEnrich (see below), overrepresented GO terms, DNA motifs, and their associated TFs were compiled into two Cytoscape compatible files that were used as network and node attribute files, and the modules are visualized with Cytoscape.
Data Output: Outputs are summarized in a text file, significant.txt, in which the gene set name is in the first column, enriched GO terms, DNA motifs, or transcription factor families are listed in the second column, and p values indicating the significance of enrichment are given in the third column. Each new enriched category is set in a new row. If a DNA motif is significantly overrepresented within a gene list (p < 0.001), it is also determined if the motif is enriched among genes significantly overrepresented for a GO term (p < 0.001). In the significant.txt file, the overrepresented GO terms are listed in the first column, enriched DNA motifs are in the second column, and p values are in the third column. TFs in the gene list (first column) that are predicted or known to bind with enriched DNA motifs (second column) are also listed in the
The significant.txt file is designed to be used as the network file for the network graphing software, Cytoscape (version 2.6.3, http://www.cytoscape.org.). The node.txt file is used as the attributes file (Cline et al., 2007). P values are also imported with the network file as edge attributes. For visualization purposes, a thicker line represents a lower p value, a dashed line represents a TF with a predicted binding interaction, and a solid red edge is an experimentally determined TF - DNA motif interaction.
References:
- Brady, S.M., Orlando, D.A., Lee, J.Y., Wang, J.Y., Koch, J., Dinneny, J.R., Mace, D., Ohler, U., and Benfey, P.N. (2007). A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801-806.
- Cline, M.S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., Christmas, R., Avila-Campilo, I., Creech, M., Gross, B., et al. (2007). Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2, 2366-2382.
- Davuluri, R.V., Sun, H., Palaniswamy, S.K., Matthews, N., Molina, C., Kurtz, M., and Grotewold, E. (2003). AGRIS: Arabidopsis gene regulatory information server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinform 4, 25.
- Gadbury, G.L., Garrett, K.A., and Allison, D.B. (2009). Challenges and approaches to statistical design and inference in high-dimensional investigations. Methods Mol Biol 553, 181-206.
- O'Connor, T.R., Dyreson, C., and Wyrick, J.J. (2005). Athena: a resource for rapid visualization and systematic analysis of Arabidopsis promoter sequences. Bioinformatics 21, 4411-4413.
- Orlando, D.A., Brady, S.M., Koch, J.D., Dinneny, J.R., and Benfey, P.N. (2009). Manipulating large-scale Arabidopsis microarray expression data: identifying dominant expression patterns and biological process enrichment. Methods Mol Biol 553, 57-77.
- Vandepoele, K., Quimbaya, M., Casneuf, T., De Veylder, L., and Van de Peer, Y. (2009). Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol 150, 535-546.
Soybean IVT Array Annotation
Sequences used for BLAST came from the Affymetrix Soybean target sequences. Sequence information can be obtained directly from Affymetrix. The Affymetrix Soybean target sequence was based on the NCBI Unigene Build 13 (November, 2003). Probe design was based on the NCBI Unigene Build as well as the Affymetrix in-house clustering algorithm. Affymetrix in-house clustering probes are designated with the prefix "GmaAffx".
BLASTX analysis was carried out using soybean target sequences searched against all Arabidopsis proteins (TAIR ATH1_pep_cm_20040228). In our BLAST analysis, we filtered and removed any results with e-value greater than e-02. We selected the top Arabidopsis hit from each BLAST result (sometimes one Soybean sequence can hit many different Arabidopsis sequence) when identifying the corresponding Arabidopsis sequence. The e-value for that hit is displayed in the annotation file. Therefore, for each Soybean probe set, there is an associated Arabidopsis annotation (if available) and the degree of homology between the Soybean and Arabidopsis sequence based on the e-value. In cases where no Arabidopsis hit was identified (~9000 Soybean probe sets did not have homology to any Arabidopsis proteins), we BLASTED the Soybean sequence against Rice Proteins (Build #2 from TIGR) and the NCBI non-redundant protein database. We annotated Soybean probe sets and did not annotate any features from H. glycines or P. sojae that are in the GeneChip.
ANNOTATION UPDATE:
Sept. 25, 2009 - We mapped individual probes to soybean predicted gene models (generated by the Department of Energy (DOE) Joint Genome Institute, Glyma version 1.01, released April 7, 2009) using BLASTN (≥ 23/25 nucleotide identity) to associate soybean array probe sets with soybean gene models. Probe sets that contain at least 9 out of 11 probes mapping to the same genomic locus are represented in the files below. Probe sets that did not meet these criteria (i.e. 23/25 nucleotide identity, ≥ 9/11 probes per probe set) were not included in the file below. We split the file into two files based on the confidence of prediction of soybean gene models (ftp://ftp.jgi-psf.org/pub/JGI_data/Glycine_max/Glyma1/annotation/highConfidence/Glyma1_highConfidence.transcriptList). Click the files below to download the association of Soybean array probe sets and Soybean gene models.
Feb. 1, 2009 - We updated the annotation of the soybean array information based on information from TAIR 7.0, TIGR, and Peking Transcription Factor databases as of October 2007. The updated information is available from the following link.
Click here to download the Soybean GeneChip annotation file (Updated Oct. 2007)
Click here to download a summary of the Soybean array annotation (Updated Oct. 2007)
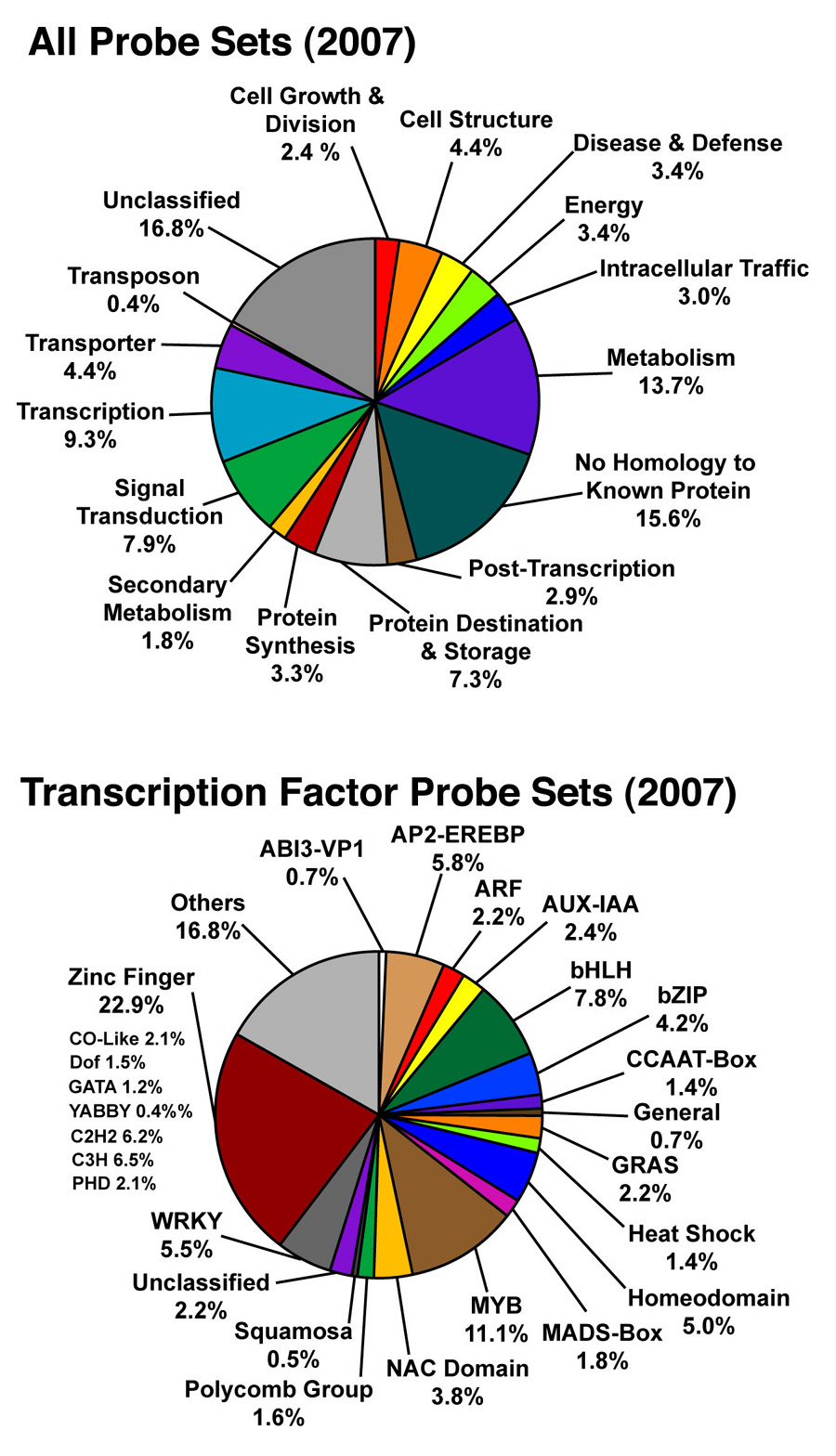
Distribution of All Probe Sets on the Soybean Array (2007)
Soybean Whole Transcript Genome Array
Motivation:
We created this Soybean Whole Transcript (WT) Array to interrogate all the genes in the genome. The first generation Affymetrix Soybean Genome array was designed by the Soybean Consortium using publicly available soybean full-length cDNAs and ESTs. The Soybean Genome array consists of 37,000 probe sets interrogating ~ 25,000 distinct genes/transcripts. The release of the whole genome sequence of soybean1 (available at Phytozome.net) allowed the creation of an array that can survey all the genes (both high and low confidence gene models) in the genome [Schmutz et al., Nature 463 pp. 178-83 (2010)].
Design:
The design of the Soybean WT array is different from the Soybean Genome array. For the Soybean Genome array, probes were selected to correspond to the 3’ end of the transcript or cDNA. However, for the Soybean WT array, probes were selected to span every exon of the predicted gene models/transcripts, if possible. This approach allows for the interrogation of the transcript (from 5’ to 3’) and can help determine exon usage in different splice variants that may be differentially expressed in specific tissues or compartments. For information regarding this array design, please check out other references from Affymetrix (http://media.affymetrix.com:80/support/technical/technotes/gene_1_0_st_technote.pdf).
Note: This array was designed for studying both Soybean and Medicago (i.e. a Legume array). There are sequences on the array corresponding to Medicago cDNAs. However, our main focus will be on the Soybean sequences on the array.
Sequence Data:
All sequence data used to design probes on the array were obtained from the Department of Energy - Joint Genome Institute (DOE-JGI) web site (phytozome: http://phytozome.net). Probes were designed from the first draft assembly of the soybean genome1 (version 1.0). The probe selection algorithm was developed by Christopher Davies and Brant Wong at Affymetrix.
Publication Acknowledgement:
The array was designed with collaboration from our lab (Goldberg Lab) and Affymetrix with advice and suggestions from other members of the soybean community, including Randy Shoemaker.
Please acknowledge the following people for the design of this array:
Goldberg Lab: Bob Goldberg, Brandon Le, Chen Cheng, Min Chen, and Anhthu Bui
Affymetrix: Gene Tanimoto, Christopher Davies, Stan Trask, Brant Wong, Eric Schell, Xue Mei Zhou, and Patricia Chan
Files for Download
[Probe Association File]
We've created a text file that correlates Affymetrix probe ID with associated probe sequence, gene and exon information, etc.
Probe Association File: [Click Here to Download]
[Soybean SENSE WT Array]
This array design is available to the general public and can be purchased through Affymetrix.
Library File: SoyGene-1_0-st-v1-rev02.zip [Click Here to Download]
Labeling Protocol: Check the Affymetrix Website for labeling and hybridization kits [Go to Affymetrix Website]
[Soybean ANTISENSE WT Array]
This array was created for our lab and is a custom-designed antisense WT array. Please use the library file and protocols listed below for this array only.
Library File: SoyGene-1_0-antisense_rev02.zip [Click Here to Download]
Labeling Protocols:
Labeling Protocol One: Nugen Ovation Pico WTA System
Click on the link to go to the product web site [Link]
Labeling Protocol Two: Ambion WT Expression Kit with Affymetrix Second Strand cDNA Synthesis
This labeling protocol is presented as is and is not regularly supported by the Affymetrix Technical Support team. This method requires an Ambion WT Expression kit, Affymetrix Fragmentation and Terminal Labeling kit, and second strand cDNA synthesis reagents from vendors provided in the attached protocol. For this protocol, you will generate cRNA using the Ambion WT Expression kit (up to Day2 Workflow, Step2). After cRNA synthesis, you will use the Affymetrix protocol (starting on page 9) to make the second cycle cDNA and terminally-labeled targets.
[Hybrididization Program]
For array wash, stain, and scan, use the fluidics protocol EuKGE-WS2v5_450 for wash and stain procedures as described in the GeneChip Expression Analysis Technical Manual (Section 2: Eukaryotic Sample and Array Processing).
Arabidopsis ATH1 Array Annotation
The Arabidopsis ATH1 array was annotated in 2003 using all the publicly available resources at the time. In order to keep up with the increasing amount of information generated within the past four years since the annotation of the ATH1 array, we decided to re-annotate the ATH1 array in parallel with the soybean genome array.
The strategy for the re-annotation of the ATH1 array is as follows:
1. We updated the descriptions for each probe set on the array using TAIR Affy array descriptions (affy_ATH1_array_elements-2007-5-2.txt). The description file was downloaded from the TAIR web site: ftp://ftp.arabidopsis.org/home/tair/Microarrays. Descriptions were based on the latest release of the Arabidopsis genome TAIR 7 (released 04-11-07).
Note from TAIR: The mapping to the TAIR7 Transcripts was performed using the BLASTN program with e-value cutoff < 9.9e-6. For the 25-mer oligo probes used on the Affy chips, the required match length to achieve this e-value is 23 or more identical nucleotides. To assign a probe set to a given locus, at least 9 of the probes included in the probe set were required to match a transcript at that locus. Otherwise, the probe set was not assigned a locus and was given the description "no match".
2. In addition to updating the descriptions for each probe set, we also updated gene ontology (GO) information provided by Affymetrix.
3. We gathered information about putative transcription factors from many publicly available TF database for Arabidopsis including:
AGRIS - Arabidopsis Gene Regulatory Information Server (http://arabidopsis.med.ohio-state.edu/)
DATF - Database of Arabidopsis Transcription Factors (http://datf.cbi.pku.edu.cn/)
RARTF - Riken Arabidopsis Transcription Factor Database (http://rarge.gsc.riken.jp/rartf/)
ArabTFDB - Arabidopsis Transcription Factor Database (http://arabtfdb.bio.uni-potsdam.de/v1.1/)
Transcription factors and transcription factor families were associated with each probe set on the array. Information obtained from points 1-3 were compiled together into an annotation file containing the 2003 ATH1 annotations. Transcription factors were automatically updated based on the information obtained from the databases in point 3.
4. We focused on probe sets that were previously assigned into the "unclassified" category. The rationale is that many of the sequences in the "unclassified" category might have update information that can be used to re-assign into a different category. Sequences previously assigned categories of "protein synthesis" or "metabolism" most likely will not change. Therefore, we first focused on re-assigning the 11,145 probe sets classified as "unclassified" in 2003.
5. After the "unclassified" category was re-examined, we decided to re-examine the entire 22,746 probe sets on the array for consistent assignment of functional categories. We sorted all the probe sets by their description and made sure that probe sets with similar descriptions are assigned the same functional category.
6. We further examined the "unclassified" category that is divided into three groups as follows:
- Unclassified - hypothetical proteins with no cDNA support
- Unclassified - hypothetical proteins with cDNA support
- Unclassified - proteins with unknown function
We obtained several files from TAIR that will distinguish the different sequences within the unclassified category. We downloaded several files from the TAIR site including:
- TAIR7_protein_coding_no_transcript_support_09_30_07
- TAIR7_protein_coding_with_transcript_support_09_30_07
- TAIR7_unknown_proteins_no_transcript_support_09_30_07
- TAIR7_proteins_of_undefined_function_03_07
- TAIR7_unknown_proteins_03_07
- TAIR7_locus_type
These files were compiled into one main table listing all the transcripts detected and/or predicted in the Arabidopsis genome. This list helps distinguish if a sequence has cDNA support, represents a pseudogene/transposon, or is unknown. These files help re-assign the probe sets into appropriate unclassified categories.
Download
The updated information is available from the following link.
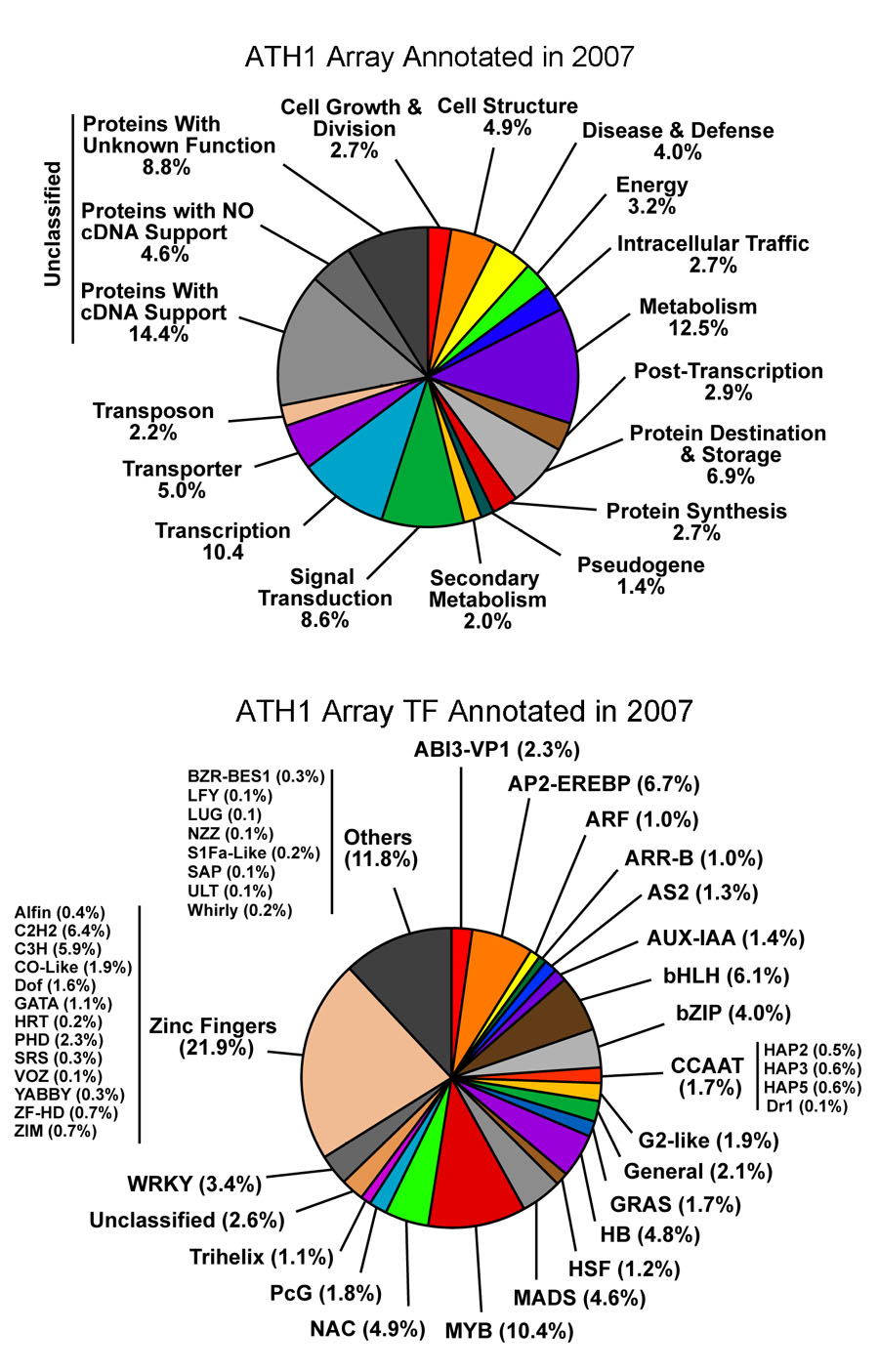
Distribution of All Probe Sets on the Arabidopsis ATH1 Array (2007)
Click the image to view larger image.
Presentations in The Past Five Years
2019
- Annual Department of Molecular, Cell, And Developmental Biology Retreat, Santa Barbara, CA (Bob Goldberg)
- International Conference on Legume Genetics and Genomics, Dijon, FRANCE (John Harada)
- Tuskegee University, AL (John Harada)
- Fort Valley State University, GA (John Harada)
- Shandong Agricultural University – UC Davis Forum, Shandong, CHINA (John Harada)
- The Plant Center Fall Retreat, University of Georgia, Athens, GA (John Harada)
- Nara Institute of Science and Technology, Nara, JAPAN (John Harada)
- International Symposium on Advanced Plant Biotechnology, Vietnam Academy of Agricultural Sciences, VIETNAM (John Harada)
- Agricultural Biotechnology Research Center, Academia Sinica, Biotechnology Center in Southern Taiwan, TAIWAN (John Harada)
- Larry Vanderhoef Memorial Lecture, National Chung-Hsing University, TAIWAN (John Harada)
2018
- Plant and Animal Genome, Legumes Workshop, San Diego, CA (John Harada)
- Addressing Public Perceptions of Modern Agriculture Forum, UC Davis, CA (John Harada)
- Soy 2018 Conference, Athens, GA (John Harada)
- Tuskegee University, AL (John Harada)
- Fort Valley State University, GA (John Harada)
- Nara Institute of Science and Technology, Nara, JAPAN (John Harada)
- Institute of Plant and Microbial Biology, Academia Sinica, Taipei, TAIWAN (John Harada)
2017
- Norm Cohn Memorial Research Symposium at Ohio University, Athens, OH (Bob Goldberg)
- Tuskegee University, AL (John Harada)
- Fort Valley State University, GA (John Harada)
- Institute of Genetics and Developmental Biology, Chinese Academy of Science, Beijing, CHINA (John Harada)
2016
- Eric Davidson Memorial Symposium at Cal Tech, Los Angeles, CA (Bob Goldberg)
- Stephen Hales Prize Invited Talk, ASPB Annual Meeting, Austin, TX (Bob Goldberg)
- Calabasas Special Speakers Series. Calabasas, CA (Bob Goldberg)
- Stephen Hales Prize Lecture. Austin, TX (Bob Goldberg)
- Soy 2016 Conference, Columbus, OH (John Harada)
- International Symposium on Agricultural Biotechnology, Taichung, TAIWAN (John Harada)
- National Chung-Hsing University, Taichung, TAIWAN (John Harada)
2015
- Center for Inquiry, Hollywood, CA (Bob Goldberg)
- The Discovery Center for Science and Technology, Thousand Oaks, CA (Bob Goldberg)
- The Seed Biology Paris Summer School, FRANCE (John Harada)
- NAIST Bio International Student Workshop Minisymposium, Nara, JAPAN (John Harada)
- National Chung-Hsing University, Taichung, TAIWAN (John Harada)
- International Symposium for Agricultural Biotechnology, Kasetsart University Kamphaeng Saen Campus, THAILAND (John Harada)
Miscellaneous Videos
These movies are best viewed in Quicktime. Click here to download Quicktime. To download the video, Mac users: Press CTRL and click on link to download video; PC user: right-click on the mouse and select download.
Seed Development Movie (2008) Developed by Brandon Le and Bob Goldberg [Download video]
Laser-capture microdissection of Arabidopsis seed compartments [Download video]
Laser-capture microdissection of soybean seed compartments [Download video]
Current & Previous Contributors to NSF Plant Genome Projects
If you have any questions or comments about this project, or data presented in this web site, please contact Bob Goldberg or John Harada.
For questions or comments about this web site, please contact Min Chen.
| UCLA | UCD | |
|
|
|
| Knockout Collaboration with Monsanto | ||
|

